Life Sentences: A Story of Discovery and Recovery
Another Afterward - What Was Killing Me? And How Was It Doing It?

When I learned I was sick, more than anything, I needed to know what was killing me. And how. So, I did some homework.
As a young man, in college, during the summer, I had a job in a bakery. Not the kind of bakery that dotted many of the corners and streets in the neighborhoods of my youth, small storefronts offering their proprietor’s special recipe for chocolate cake with vanilla cream frosting or cannoli. It was an industrial bakery. In the summer, when I worked there, during my breaks from college, the factory—for in my mind it was more factory than bakery—operated full time, three shifts of eight hours. Brought in so the factory could run the third shift, my youthful colleagues and I were dispersed among the regular workers, trained at first to do the easier jobs. Then, we were given progressively more challenging, and perhaps less desirable, duties. I say perhaps because my first assignment, which I viewed as highly undesirable, involved standing at a conveyor belt watching hamburger rolls enter a slicing machine. My job was to ensure there were no backups on the belt, which would clog the slicer thereby ruining large quantities of rolls. I also watched for unexpected gaps on the belt. That meant a loss of production, which resulted in missing the shift’s quota, causing grief and hard work for the following shift’s workers who would have to make up the gap. It was summer and burgers were in high demand. To feed this appetite, my factory baked hamburger rolls all day, every day. Monitoring the rolls entering the slicer was the least skillful job in the factory. All newbies started there.
My bakery job has nothing to do with my genetics or my disease, but in searching my familiar world for a metaphor for my condition to help me answer the “what/how is this happening to me” question, not metaphysically but physically, the bakery cum factory is a decent one. In the J. J. Nissen Bakery that made hamburgers rolls all day, every day each summer in Central Falls, RI, the smallest problem, or malfunction could have important consequences, some potentially disastrous, for roll production. Ingredients may have been wrongly measured at the dough mixer causing the rolls to be flatter than necessary. The oven temperature may have been improperly calibrated causing the rolls to be undercooked or burned. Or a young college student, during the third shift at 3a.m., after going to the beach instead of sleeping the previous day, might incorrectly load a platen, maybe using a six instead of a nine, resulting in batches upon batches of rolls priced below cost, and in lost profits. Somewhere, bookkeepers know there is a problem, but they do not know why. The source eludes them. Meanwhile, the perpetually exhausted college student continues to load the six instead of the nine. His error becomes his norm. The mistake might spread to other shifts; it becomes a creeping, degenerative force within the factory. Eventually, and prematurely, owing to slow, cumulative, uncontrolled financial loss, the bakery will go out of business. Fortunately for hamburger roll bakeries, or any production facility for that matter, good basic accounting, or “continuous improvement,” or “leaning” could root out the collegian’s error and right the listing ship before it sank. The human factory is infinitely more complex than any human-developed production space, however, and the quality control gurus of the body—medical doctors and scientists—are far from capable, yet, of fully understanding and correcting the errors that arise. My health is like the factory, and one of my genes is the college student. It is called RTEL1. It will cause my premature death.
Having dabbled in the world of science and math as a student and professional, I know the use of the term “infinitely more” is always hyperbole. The scope of our genetic mixture, however, lends itself to such hyperbolic statements, revealed by a look at the numbers. Each of us has 23 pairs of chromosomes, one set from each parent, and each of the tens of trillions of cells in our body has these same 23 chromosome pairs. Chromosomes vary in size from pair to pair, but each is part of a six-billion rung ladder called DNA, or Deoxyribonucleic Acid, usually illustrated by the familiar “double helix.” To picture the actual chromosomes, imagine cutting the six billion rung ladder into 46 smaller ladders of 23 different lengths. Ladders are twisted, the double helix, and then each is wrapped around a “protein” ball four times. After the fourth wrap, imagine moving onto the next ball, until reaching the end. This ladder-and-ball construction is then spiraled, giving us the physical form of a chromosome. Inside the cell, these chromosomes are like noodles, 46 pieces, in a bowl of broth, and when necessary for cell replication, the same-sized noodles pair with one another. To mix metaphors, the longest noodle is 250 million rungs long, the smallest is 47 million, and the noodles (i.e., chromosomes) are numbered one through 23. A gene is a subset of a chromosome, a certain number of rungs, each varying in length from 800 rungs to more than two million. Chromosome number 20, where the gene RTEL resides, is 65 million rungs long. The RTEL1 gene is about 38,000 rungs long. That makes it about one ten thousandth of one percent of the six billion rung human DNA “ladder.”
A gene holds instructions, a recipe or computer code, for our growth, the division of our cells, and our operation, how our body works. Continuing the ladder analogy, each rung has writing on it. Some of the writing is nonsense or unintelligible, while other rungs, when viewed in sequence, contain instructions for accomplishing critical tasks. Either, the instructions provide the directions for replicating the ladder, identically. Or, the instructions provide a recipe for making molecules, proteins, that operate our body. The complete set of instructions is called the human exome; the smaller sequences of instructions on each rung or set of rungs are called exons. The exome is less than two percent of the total genetic “writing” space available in our body. It is like a piece of paper of random letters except for a small space on which a recipe is written. The random letters on the paper, with equally obtuse (introns) or vague (Untranslated Regions) names, contain no obvious instructions. These random bits are not unimportant, but scientists do not fully understand their role yet. In RTEL1 there are 35 different exons, or sequence blocks of instructions, comprised of about 3700 rungs of the 38,000-rung RTEL1 section of the “ladder.” The important instructions in the RTEL1 gene, therefore, are one hundred thousandth of one percent of human DNA (the genome).
If we are born with mistakes in our genetic instructions, or if they do not remain intact as we grow, good or bad things can happen. Keeping with the ladder metaphor, imagine that each rung on our ladder is made by adjoining two different materials, each encoded with instructions that can be read sensibly only when matched with a proper second half. These two materials, and their attendant instructions, are known to work well together; they have been tested. Sometimes, however, in the reproduction, or copying, of the ladder, changes occur to the material or the instructions. Perhaps a different material pairing is discovered, and the ladder and its instructions are improved. The rung is better. Or imagine a material change is made resulting in a weaker rung, the instructions written on that rung sounding like nonsense, or resulting in a break in the ladder altogether, prohibiting further replication. These changes are called mutations and they occur frequently. Sometimes mutations are beneficial or may have been essential for the survival of a species: one recently discovered mutation, for instance, confers on its holders resistance to HIV; and one evolutionary theory claims humans’ ability to see in trichromatic vision was probably the result of a mutation millions of years ago giving us a survival advantage in a world of mostly dichromatic animals.
Other mutations can be dire. In the RTEL1 gene, change does not seem to be good. In my case, I was born with a single rung of material, out of a total of more than 3700 on my RTEL1 gene, that has incorrect instructions, a recipe error. Again, half of one rung, out of a total potential of 6 billion rungs (3 billion “matching” rungs from each parent), a hundred millionth of one percent (.000000001) chance, is causing my premature death. The numbers are staggering. Not infinite, of course, but staggering.
My “Reaper” gene, as I prefer to call it, is a regulator, its full name being Regulator of Telomere Elongation Helicase-1. A helicase is an enzyme with two functions: it unwraps the double helix (our twisted ladder) facilitating the production of new “ladders”; and it reads the instructions encoded on the ladders’ rungs. An enzyme is a protein that catalyzes a chemical reaction. And the “-1” means it’s the first gene of its kind to be discovered in this category. The RTEL1 gene, then, provides instructions for the making and operation of a protein that unwinds our DNA to facilitate the regulation of the elongation of a telomere during chromosome (and cell) replication. My telomeres are not being elongated properly during replication, a problem whose importance cannot be overstated. And though we know, in me, RTEL1 is not functioning properly, science still does not know how the helicase operates (or fails to operate) to cause telomere shortening—only that it does.
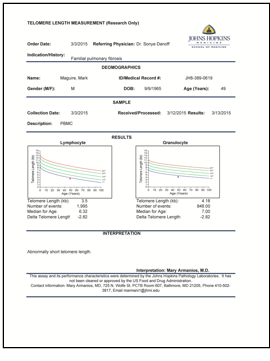
My telomere test results showing mine (the dot) are less than one percentile of normal telomeres.
Telomeres are the ends of chromosomes and are the key to a long life, unless they are the key to a short one. And either might be the case. Long telomeres are good; short telomeres and too-long telomeres are bad. Telomeres, it seems, have a “Goldilocks” zone, a “just-right” length at which we live a long healthy life, unless we are met with other catastrophic circumstances – the ubiquitous “getting-run-over-by-a-bus” scenario is always looming. This is all a bit of embellishment, but not too much. Recently, a cottage industry has begun to emerge surrounding the testing and maintenance of telomere length. Commercials have appeared on television. Books are being published. A Nobel laureate gave a TED talk on this topic. There are startups based on telomere health. Provide a DNA sample and a company can measure one’s telomeres and then provide therapeutic approaches either to lengthen them or maintain their current healthy length. Academic papers are emerging with studies and analyses claiming stress and early childhood trauma shorten telomeres, the implication being reversal or avoidance of these stimuli will lead to a healthier life. I cannot take issue with any recommendation to avoid stress or early trauma, but my doctors did not disagree when I characterized the entire “telomere industry” as modern-day snake-oil salespeople. My doctors’ research indicates that human telomere length fits into well-defined normal range and factors that shorten them are genetic in nature. Claims regarding the effect of external stimuli, they assert, may be dependent on testing methodologies and the associated error margins. That being said, this is an exploding field, and much is possible. Genetic manipulation to lengthen telomeres is emerging and may one day be commonplace. As a cautionary note, however, before hastily mixing a potion or genetically engineering our bodies to lengthen our telomeres, one should be aware that too-long telomeres have been associated with cancer risk. The law of unintended consequences is alive and well. None of this science is certain yet. What is certain is that my genetics lead to short telomeres and that short telomeres lead to problems.
To understand what was happening to my telomeres owing to my RTEL-1 mutation, my ninth-grade biology course was not enough. It needed supplementing. I had to delve further. The whole thing is a bit ponderous, but also wondrous.
Our growth and health depend on cell replication, the cell’s creation of a perfect copy of itself by duplicating, among other things, each chromosome. Of critical importance is that the chromosome does not lose any information during replication. Our body contains “quality control” mechanisms that will cause a cell to self-destruct if chromosome information is incomplete, which can happen if the end of the chromosome becomes truncated, or if the chromosome breaks somewhere in the middle. Consider the ladder again. It has been twisted and wrapped and spiraled. Copying the ladder in this state is dubious. Recall, the rungs have information on them – how does one confidently read each rung if the ladder is still twisted, wrapped, and spiraled. Amazingly, life has evolved an intricate machinery to accomplish chromosome or DNA replication. There is an un-doing: un-do the spiraling, un-do the wrapping and un-do the twisting. The spirals and warps and twist were very useful for storing our DNA in a cell but not so useful for duplicating it. With a relatively straight ladder, one clearly sees each rung, facilitating the copy, for which evolution developed a complex, and messy and some might say inelegant, method: our bodies are programmed to cut the ladder through the center of each rung and build an identical new matching half for each of the original halves, as the cutting happens. While being constructed, the new halves are attached to the original halves. Efficaciously, the replication process (cutting and copying) begins near the center of the ladder and happens in two directions, reducing production time. At the end, we have a perfect copy of the ladder, all 46 ladders in fact, ready to be twisted, wrapped, spiraled, and paired to make a chromosome and a new cell.
Unfortunately, biology must deal with the complexity of chemistry, which necessitated evolution’s creation of telomeres, to ensure sound replication. Because of the laws of chemical bonds, the copying machinery, working in two directions, cannot produce both new halves of the ladder at the same time; there is a leading half and a lagging half. The leading side proceeds normally—that is, continuously—without any trouble, while the lagging side has to stop and start the copying process every 1000 rungs, or so, and “glue” each segment back together. Incredibly, both halves get copied until the very end of the chromosome when the lagging side cannot start the last 1000 rungs of the ladder because the leading side is finished, causing the need to shorten the newest, copied ladder. Using a simpler, albeit cruder, analogy, imagine the work clock in a ladder production factory stopping after the “lead” crew has completed its half of the ladder, but the “lagging” crew still has ten rungs to go. Science has not yet given us the ability to reorient, or mix, the crews so they perform equally, nor can we stop the “lead” crew until the “lagging” crew catches up. Our only solution is to shorten the ladder by ten rungs (the rungs that would be incomplete when we paired the two crews’ pieces together), thereby preventing an uneven, or tilted, product. By doing this, we risk selling too-short ladders, or ladders that do not perform critical functions—the regulation of our growth and health gone amiss. If it happens that information is lost, we must discard the ladder as trash—cells that do not have all their critical information are designed to self-destruct, to senesce, to die. Remedy becomes disease.
Telomeres are biology’s answer to this problem, which scientists call the “end replication” dilemma. Telomeres are extensions on our ladder, comprised of rungs containing nothing but meaningless information – a repeating nonsensical sequence of molecules called nucleotides – the sole purpose of which is to give leeway for the lagging half when it fails to copy the last rungs of the ladder. The new ladder, although truncated at the ends, does not lose the important instructions that are found on the ladder—the shortened ladders are still tall enough for the job. Following this logic, shortening the ladder after each production run only delays loss of critical instructions, it does not prevent that outcome. Eventually, there will be no more telomere extension and we are again faced with the “end replication” dilemma – in fact, this is what happens. Replication, for the vast majority of our cells, has an upper limit and when that limit is reached, cells senesce, or die. The limit, which is called the Hayflick limit and correlates with, or depends on, our telomere length, and is on the order of 50-60 replication cycles per cell, permits us a long, happy life, notwithstanding the “bus scenario.” Aging is the result of our trillions of cells reaching their replication limit.
A small subset of special cells in our bodies are less subject to cell replication limits. Stem cells can continue their replication with less degradation by using an enzyme called telomerase. During each replication telomerase acts to lengthen telomeres, adding the nonsense nucleotide molecules. In this context, one can understand the focus and success of companies attempting to “bottle” telomere extension “potions” as extending telomeres appears, in this telomere-based model of cell death, to slow the aging process, extending life. Alas, there is no scientific proof, yet, that medical therapies can accomplish this goal; there is no telomerase in a bottle, but with emerging technologies, it may be possible someday. So, stem cells and telomerase exist throughout our bodies, but not in sufficient numbers and quantity to overcome the evolutionarily programmed cell-aging process, which, despite its rather dire consequences for our lives, must be recognized for its positive attributes. Uncontrolled cell growth is cancer, and data shows that telomerase is overexpressed in cancerous cells, meaning, perhaps, that in cancer, natural cell death has been forestalled by a continual lengthening of telomeres. If telomere-lengthening therapies are developed, caution about the correlation between long telomeres and cancer needs consideration.
As the end of chromosomes, telomeres are constantly under attack as a faulty break, or “loose end,” a second critical problem solved by telomere evolution. Just as nature abhors a vacuum, our bodies abhor chromosome loose ends. We have developed a “search and destroy” defense mechanism to eliminate the possibility of broken DNA replicating itself and creating a monster, or mutant, chromosome, if you will—a loose end of a chromosome is subject to fusion with another loose end, thereby creating junk material that our bodies discard as irrelevant to our genetic growth. This is referred to as the “end protection” problem. Some mutations are successful, others not so much, but our replication biology does not have predictive capability—it does not know if a mutation will be good or bad, it just is. Rather, our biology is programmed to copy what exists. Lest they infiltrate the replication machinery, obstacles to exact copies of DNA, stray loose ends in this case, must be destroyed. [The “search and destroy” mechanism is quite effective for loose ends, but not so much for mutations that occur in the middle of a DNA strand.] But telomeres, by definition, are the loose ends of chromosomes. So how does a telomere outwit a defense mechanism designed to recognize loose ends and eradicate them, fusing them with other loose ends, and committing them to the oblivion of useless genetic material, never to be copied? Ingeniously, evolution has furnished telomeres with a disguise. A telomere evades the loose end surveillance pathway with the help of the shelterin complex, a set of proteins that bind to telomeres, hiding the DNA from the surveillance machinery. The complex is akin to a set of tinkerers who have learned through millions of years of evolution exactly what disguise to put on the telomere to protect it against the loose-end surveillance system. Exactly how this happens—what the disguise is—remains unclear. According to one proposed model of this phenomena, with the help of the complex, the chromosome loops back on itself and tucks its end into an earlier part of the telomere. The entire structure resembles a “lariat” with the larger loop called the “telomere-loop” and the smaller connecting loop called the “displacement loop.” Nature’s “loose-end” hunters still look for the end of the telomere, but the loop structure is sufficient to throw the hunters off the trail. Successful in their effort, the tinkerers have protected the ends of chromosomes from being identified as problems, preventing that chromosome from being discarded, preserving our life’s instructions.
Telomeres and their associates, the shelterin complex, then, solve two problems: the “end replication” problem and the “end protection” problem. One solution protects against critical genetic material being lost, which would cause cell replication to stop prematurely, not reaching the 50-60 cell division limit that leads to a full life expectancy. The other protects against quality control mechanisms that would identify the end of the chromosome as a fault, causing the chromosome’s destruction and a similar end, premature cell death. These two solutions are not complementary; in fact, they conflict. Proper cell division requires the undoing of spirals and wraps and twists, of which the telomere “lariat” is a part. As mechanisms of untangling DNA proceed, they are confronted with a “knot” in the DNA strand, put in place by the very telomere now being copied. The telomere has knotted itself into a loop for protection. Unless the knot in the loop is undone, replication will stop. The strand will be cut. The resulting telomere will be too short. The cell will distribute a directive to stop reproducing, to senesce. RTEL1, scientists believe, is critical for undoing the knot.
I take some issue with the genetic scientists who named RTEL1. In standard English, the primary definition of the word “elongation” is to lengthen something. To the best of my knowledge—admittedly quite limited in this regard—RTEL1 is not elongating telomeres, or even regulating their elongation. Telomeres, in all but a very small subset of our cells—those hematopoietic stem cells for example—get shorter over time whether RTEL1 is working properly or not. In biological terms, however, elongation can refer to the process of adding the next sequence of information during replication—adding the next rung to the new half of our ladder. In this sense, RTEL1 helps regulate the copying of our telomeres during cell division. More specifically, based on the loop theory, it ensures telomeres are untangled, for the replication activity to function smoothly and for cells to reach their natural replicative limit—that the shelterin complex knot is not perceived as the end of the chromosome. If RTEL1 does not function properly, cell division is compromised.
Genetically speaking, my mutation is a single nucleotide change, a heterozygous missense mutation in my germline. A fancy way to say one of my parents gave a bad copy of it to me. A single molecule in one of the two copies of Chromosome 20 is wrong. In me, a Guanine molecule has replaced an Adenine molecule at the 62,324,601st base pair on Chromosome 20 – one base pair out of 6 billion pairs. Most certainly I have other mistaken base pairs, but to date, none have revealed themselves, and none are killing me, to my knowledge. The result of this single nucleotide polymorphism, as it’s known, is that the exon associated with that base pair, that small instruction on our imaginary adder, provides directions, a codon, to make a Glutamine amino acid instead of an Arginine amino acid. Amino acids make the proteins that make the molecules that untie the knots. It’s all esoteric until one is progressively short of breath. My mistake occurs near the end of the RTEL1 gene’s encoding instructions, after creating nearly 1000 of the 1300 intended amino acids. Unfortunately, this minor error stops the remainder of the instructions from being transmitted. I was almost there. Properly encoded, the amino acids would have created one or more proteins that would untangle the telomere. Instead, my telomere “knot” is not undone, the replication process stops, my telomeres get shorter and some of my cells senesce.
Scientists speculate that cell senescence, the inevitable outcome of shortening telomeres in most of our cells, is a key mechanism for fibrosis generally, and pulmonary fibrosis specifically. Pathogenic is the word used. Senescence is pathogenic to fibrosis. Speculation is the best we have at this point, but the statistics are undeniable. Nearly half of patients with pulmonary fibrosis have been shown to have a defect in their telomere pathway, and that number increases with further research. Some scientists believe the number may continue to grow. My genetic mistake was a one-in-a-few-billion chance mutation, but mutations in RTEL1 come in different flavors and there are other genes, with other potential mutations, that are also part the human “telomerase” complex, the term given our body’s machinery for telomere maintenance. It may be proven that the sum of all telomere pathway defects[1] amounts to the key feature of all pulmonary fibrosis cases, but even with overwhelming statistical evidence, a precise explanation of the biological process remains unknown.
Fibrosis is the accumulation of excess “fibrous” tissue in response to an injury. A scar on one’s skin is a good example. In pulmonary fibrosis, the scar, or thickening of connective tissue, occurs around the interstitium of the lung, which is why pulmonary fibrosis fits into a group of diseases called Interstitial Lung Diseases. Interstitium is the space between our airways and our blood vessels, the bit through which oxygen and carbon dioxide flow; this flow happens as we breath. Lungs are not solid. Picture a large upside-down tree, before it blooms, with many branches and stems, each ending in a set of buds, 300 million in total. In each of our lungs, we have dual complementary trees. One tree is our airway, another is our blood vessel; they are side-by-side, separate until each reaches the tips of the branches, where the buds are. Instead of buds, in our lungs, the blood-vessel tree has tiny nets, or capillaries, that wrap around the airway tree’s buds, or alveoli. Between the net and the bud is interstitial space, in which several different kinds of cells are busily going about their biological duty. Some of the simplest cells comprise a lining, called epithelial cells in the alveoli and endothelial cells in the capillaries. Thin enough and porous, these cells permit the passing of oxygen and carbon dioxide molecules through their cellular walls, a gas exchange, or breathing. Other cells are more complex. One secretes a mixture that coats the surface of the alveoli, ensuring it remains flexible and porous, and it also helps rebuild the alveoli lining when necessary; one watches for and removes undesirable material breathed into the alveoli; and one is charged with maintaining the connective tissue, or support structure, of the lung. There are others. How some, or all, of these cells interact, or fail to perform, to cause fibrosis is unknown. As statistical evidence builds for the link between short telomeres and fibrosis, however, cell senescence gathers evidentiary momentum as a primary pathogenesis of pulmonary fibrosis. In this theory, alveolar epithelial cells fail owing to a DNA damage response as a result of the cells’ short telomeres. These cells have natural damage response functions aimed at maintaining what scientists refer to as homeostasis, or balance, allowing a consistent flow across the epithelial membrane. In this case, of oxygen and carbon dioxide molecules. This damage response mechanism is always active, constantly alert for senescent cells, those that have reached their replicative limit, and, working in conjunction with the cells that rebuild our lungs’ epithelial lining, adjusts the surrounding connective tissue in order achieve homeostasis. Unfortunately for patients afflicted with short telomeres, an overabundance of senescent cells can cause an over-reactive damage response and the formation of excess connective tissue, a fibrous lesion. These fibrous lesions inhibit the flow of molecules across the alveoli membrane. Oxygen stops flowing. Suffocation ensues.
This is not hyperbole. Unless I choose the other, potentially more risky journey of getting a lung transplant, I will die of slow suffocation. Eventually, because of this scarring in my lungs, my alveoli will not exchange oxygen and carbon dioxide with my capillaries. My organs will fail but, gratefully, I will lose consciousness before that happens. Medicine, should I choose, will attempt, first, to offset this decline with supplemental oxygen, using a differential pressure to force oxygen through the alveoli membrane, but even that will fail as there will be no interstitial membrane space through which oxygen can pass. Ultimately, there is the option for extracorporeal membrane oxygenation, or ECMO, which extracts blood from the body, oxygenates it, and replaces it. ECMO is usually reserved to bridge the gap to transplant as long-term use has some unfortunate potential consequences, brain death among them. Despite progress being made in this field, I will avoid this path.
This “RTEL1-short telomeres-cell senescence” explanation has the virtue of being sensible, but the entire theory is not without shortcomings and serious questions. Most important of these are whether and to what extent environmental factors couple with short telomeres to effectuate a damage response in the lungs. Avoidance of any such environment could be a critical lifestyle choice. There are non-senescent theories of lung fibrosis as well. Are these other theories consistent with the statistics? Are short telomeres in pulmonary fibrosis patients a coincidence, or do they play an unknown role in the other pathogenic theories of lung fibrosis? I am hopeful that scientists and doctors will sort out an answer. It may be too late for me, but my two children, if they are afflicted, could benefit.
[1]Some in the medical field dispute the use of “all” in the prior sentence, believing that the telomere pathway will never account for all pulmonary fibrosis cases, that other causes will be identified. I would argue that that belief would result in a very inelegant explanation: more than one cause for the same ailment. It is more likely, I believe, that science simply has missed a more fundamental cause. Alternatively, medicine is identifying two different ailments as one.